Main Analysis Options
CloudBlast
CloudBlast is a cloud-based Blast2GO PRO Community resource for massive sequence alignment tasks. It allows to execute official NCBI Blast+ searches in a dedicated computing cloud without additional setup completely independent from the NCBI servers. CloudBlast is a highly efficient, secure and cost optimized solution for your analysis. CloudBlast allows you to perform Blast searches for tens of thousands of sequences within a few days against a large collection of protein databases. Each sequence alignment performed in the system consumes a certain amount of computation time (translated into Computation Units) depending on the sequence length, the used Blast algorithm (blastx, blastp) and parameters used. The smaller the database, the more sequences you can analyze with a certain amount of Computation Units.
To get an idea of the consumption of these units here is an example:
With 3.000.000 Computation Units you would be able to blastx close to 500.000 sequences against the vertebrate NR-subset. A Blast search against the entire NR database, the largest protein database available, should allow you to process approx. 35.000 sequences (with an average length of 800nt per sequence). The difference in consumption can be explained due to the different size of the protein target databases - which result in a significant reduction of the amount of computation time required for a given amount of sequences - which speeds-up your overall analysis. These numbers are only orientative and change over time due to the increase of sequences available in public database collections.
GO Mapping
GO Mapping is the process of retrieving Gene Ontology terms associated to the hits obtained after a BLAST search. To run GO Mapping, select one or various data-sets, which contain blasted sequences and execute the mapping function. When a BLAST result is successfully mapped to GO terms, these will come up at the GOs column of the main sequence table. Assigned GOs can be reviewed in the Blast Result or Mapping Result, both available from the context menu (right-click a sequence). Successfully mapped sequences will turn green. Blast2GO performs different mapping steps to link all BLAST hits to the functional information stored in the Gene Ontology database. Therefore Blast2GO uses 2 public resources provided by the EMBL-EBI and Gene Ontology Annotation project. First, protein Blast hit identifiers (protein accessions or GeneInfo identifiers) are translated to UniProt protein identifiers. The obtained protein ids lead are annotated with GO terms, which are retrieved together with their evidence codes etc. The GO database contains several million functionally annotated gene products for hundreds of different species. All annotations are associated to evidence codes which provide information about the quality and type of origin of this original functional assignment.
Important:
The GO Mapping step only works reliable when BlastX or BlastP have been performed earlier to find functional homologies.
GO Annotation
This is the process of selecting GO terms from the GO pool obtained by the GO Mapping step and assigning them to the query sequences. GO Annotation is carried out by applying an annotation rule (AR) on the found ontology terms. The rule seeks to find the most specific annotations with a certain level of reliability. This process is adjustable in specificity and stringency. For each candidate GO an annotation score (AS) is computed. The AS is composed of two additive terms. The first, direct term (DT), represents the highest hit similarity of this GO weighted by a factor corresponding to its EC.
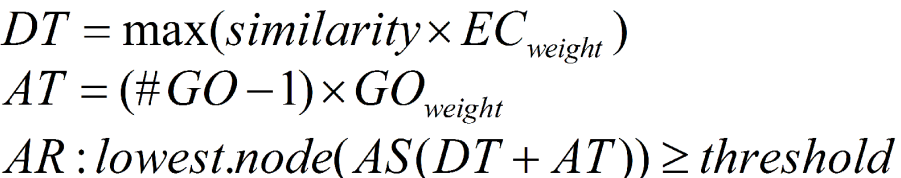
Figure 1: Annotation Rule
The second term (AT) of the AS provides the possibility of abstraction. This is defined as annotation to a parent node when several child nodes are present in the GO candidate collection. This term multiplies the number of total GOs unified at the node by a user defined GO weight factor that controls the possibility and strength of abstraction. When GO weight is set to 0, no abstraction is done.
Finally, the AR selects the lowest term per branch that lies over a user defined threshold. DT, AT and the AR terms are defined as given in the following figure.
To better understand how the annotation score works, the following reasoning can be done:
When EC-weight is set to 1 for all ECs (no EC influence) and GO-weight equals zero (no abstraction), then the annotation score equals the maximum similarity value of the hits that have that GO term and the sequence will be annotated with that GO term if that score is above the given threshold provided. The situation when EC-weights are lower than 1 means that higher similarities are required to reach the threshold. If the GO-weight is different to 0 this means that the possibility is enabled that a parent node will reach the threshold while its various children nodes would not. The annotation rule provides a general framework for annotation. The actual way annotation occurs depends on how the different parameters at the AS are set.
- E-Value Hit Filter. This value can be understood as a pre-filter: only GO terms obtained from hits with a greater e-value than given will be used for annotation and/or shown in a generated graph (default=1.0E-6).
- Annotation Cut-Off (threshold).The annotation rule selects the lowest term per branch that lies over this threshold (default=55).
- GO-Weight. This is the weight given to the contribution of mapped children terms to the annotation of a parent term (default=5).
- Hsp-Hit Coverage CutOff. Sets the minimum needed coverage between a Hit and his HSP. For example a value of 80 would mean that the aligned HSP must cover at least 80% of the longitude of its Hit. Only annotations from Hit fulfilling this criterion will be considered for annotation transference.
- EC-Weight. Note that in case influence by evidence codes is not wanted, you can set them all at 1. Alternatively, when you want to exclude GO annotations of a certain EC (for example IEAs), you can set this EC weight at 0.
A detailed explanation of the GO-Evidence-Codes can be found here: http://www.geneontology.org/GO.evidence.shtml.
Successful annotation for each query sequence will result in a color change for that sequence from light-green to blue at the Main Sequence Table, and only the annotated GOs will remain in the GO IDs column. An overview of the extent and intensity of the annotation can be obtained from the Annotation Distribution Chart, which shows the number of sequences annotated at different amounts of GO-terms.
InterProScan
The functionality of InterPro annotations in Blast2GO allows to retrieve domain/motif information in a sequence-wise manner. The processed sequence have to contain a valid sequence string. This is not the case when your Blast2GO project has been created via blast result import. Many InterProScan families are directly related to certain biological functions and linked to the corresponding Gene Ontology terms. Functional information obtained via the algorithm that form part of the InterProScan family can in a subsequent step be added to the information already available for your sequence data. To merge domain based GO terms to the once obtained via the blast based annotation step the "Merge InterProScan Results" function has to be called. If this step is omitted the GO terms obtained via the InterPro are not added and combined with the already existing annotations. Result details can be viewed through the Single Sequence Menu.
Important:
You have to provide a valid email address to be able to run the InterProScan at EBI.
GO-Slim
What is a GO Slim?
(Ref: Gene Ontology website, http://geneontology.org/page/go-slim-and-subset-guide)
GO slims are cut-down versions of the Gene Ontology containing a subset of the terms in the whole GO. They give a broad overview of the ontology content without the detail of the specific fine grained terms. GO slims are particularly useful for giving a summary of the results of GO annotation of a genome, microarray, or cDNA collection when broad classification of gene product function is required. GO slims are created by users according to their needs, and may be specific to species or to particular areas of the ontologies. GO provides a generic GO slim which, like the GO itself, is not species specific, and which should be suitable for most purposes. Alternatively, users can create their own GO slims or use one of the model organismspecific slims integrated into the GO flat file. Please email the GO helpdesk for more information about creating and submitting your GO slim.
To get a better understanding of what GO Slim does in practice and how it works, here (Figure 2) is a small visual example.
Imagine figure 2a to be the subset of GO terms called GO Slim, figure 2b shows a data-set with GO 6,9 and 10 annotated. The GO Slim methodology will pull up the 3 annotated GOs as follows:
- 6 > 1
- 9 > 4
- 10 > 5
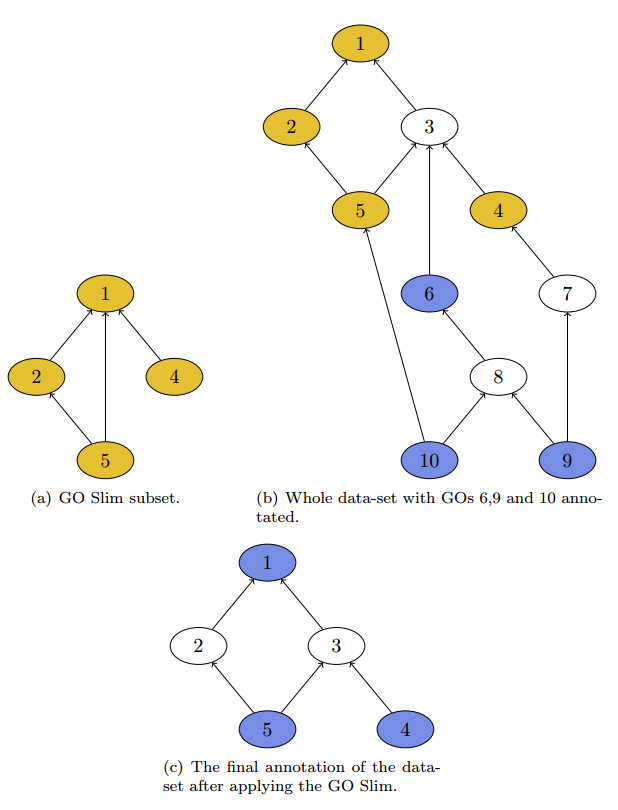
Figure 2: This shows an example of GO Slim in practice, each node represents one GO. White stands for normal, yellow for GO Slim and blue for directly annotated.
The result is shown in figure 2c. Keep in mind that this would be a data-set containing various sequences, because one sequence that has annotated GO 1 and 4 would remain only with GO 4 because of the true-path rule.
In the application our GO Slim subset is represented by a file with the extension .obo, this file contains all GO nodes and their hierarchical structure. The Gene Ontology Consortium provides various GO Slims that can be used and accessed directly from within the application. To select a predefined GO Slim, select Obo file from GO-Website and select your preferred file, it will then be used in combination with the currently selected obo file under Edit > Preferences > General > Blast2GO Data Access Settings > Change Settings > Obo File selection at the bottom. The latter file contains the whole set of Gene Ontology terms.
If the user wants to experiment and to try something separate, he can go for Custom obo files and select the two obo files by hand. Keep in mind that the GO Slim file has to contain a real subset of GOs, otherwise the result is undefined.