Quick Start
This section provides a quick run-through of a basic functional annotation process done with Blast2GO. More detailed descriptions of the different analysis steps and more advanced features are described in the remaining sections of this documentation.
- To start an annotation process load a Fasta sequence file:
Import > Standard Import > File Type: Fasta File.
You can also add an example dataset to your Navigation Area from: Toolbox > Blast2GO > Miscellaneous > Example Data
This dataset contains 100 sequences as plain sequences to start with. - Blast your sequences:
You have basically 3 options here.- Blast2GO's CloudBlast feature from the Blast2GO Toolbox. You first need to convert your Fasta sequences into a Blast2GO Project:
Toolbox > Blast2GO > Manage Projects > Convert to Blast2GO Project. - Genomics Workbench built-in Blast functionalities. Please see: BLAST at NCBI in the Workbench Help.
- Import Blast results with Toolbox > Blast2GO > Import > Import Blast Results.
The GO Mapping only works if the sequences have been blasted with an adequate Blast program. Be sure to run blastx or blastp, since you need to get protein IDs. - Blast2GO's CloudBlast feature from the Blast2GO Toolbox. You first need to convert your Fasta sequences into a Blast2GO Project:
- Convert Blast results into a Blast2GO Project (skip this if you already have a Blast2GO Project e.g. if you used CloudBlast):
Go to Toolbox > Blast2GO > Manage Projects > Convert to Blast2GO Project to convert your Blast results into a Blast2GO Project. Here you can select Multi Blast results created with the CLC Workbench. - Perform Gene Ontology Mapping:
Go to Toolbox > Blast2GO > Mapping to start the process. Mapped sequences will turn green, visualize your results once finished: Toolbox > Blast2GO > Statistics > Mapping. - Annotation:
Go to Toolbox > Blast2GO > Annotation to run the annotation step and observe that successfully annotated sequences will turn blue. - Generate Statistics Charts:
Once the annotation process has finished we can generate all the different statistics charts from: Toolbox > Blast2GO > Statistics. - Modify Annotations:
To manually modify annotations of a single sequence, right-click on one of the sequences from the Blast2GO sequence table and select Change Annotation and Description. To summarize the functional content of a dataset you can run a GO-Slim reduction (Toolbox > Blast2GO > GO-Slim). - InterProScan:
To complement the Blast homology inferred annotations with domain-based annotations, run an InterProScan search. Go to Toolbox > Blast2GO > InterProScan. This step is recommended to improve the annotation outcome. Once InterProScan results are retrieved, use Toolbox > Blast2GO > Merge GOs from InterProScan to add the GO terms obtained through motifs/domains to the current/existing annotations. - Export Results:
The Blast2GO plugin offers various possibilities to export data through the workbenches Export and Graphics function. Some of the most important are:- annot-file: The annot file is the standard format to export GO annotations. It is a tab-separated text file, each row contains one GO term.
- b2g-file: The standard Blast2GO project file. This file can also be opened with the standalone Blast2GO application.
- Sequence Table: A tab-separated text file containing all the information given in the Blast2GO sequence table, available from the sequence editor's sidepanel.
- GAF 2.0: A tab-separated text file of the functional information in the Gene Ontology annotation file format. The content of this format can also be viewed within the workbench via the Annotation Table function from the toolbox.
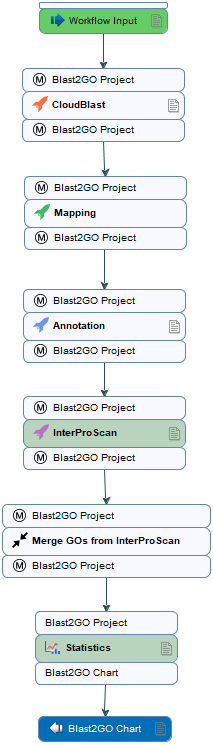
Figure 1: The workflow from the example data shows a similar scenario as described above. The work-flow accepts a Blast2GO project as input (plain Fasta sequences). It proceeds with CloudBlast, GO Mapping, GO Annotation, InterproScan, merges the annotations obtained through Blast and Domain searches and generates several charts on the way.